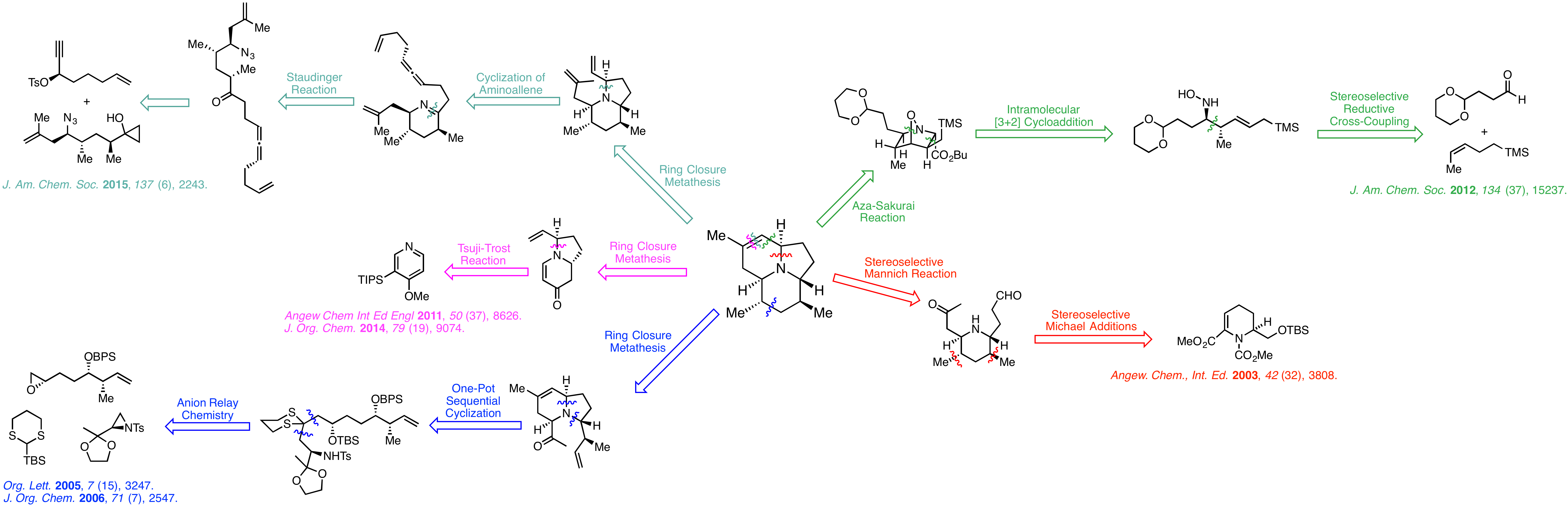
Synthesis of 205B
从蛙类Dendrobates pumilio皮肤中提取的天然产物(−)-205B,自1998年结构得到确认之后,其氮杂三环的骨架受到化学家广泛的关注。2003年Toyooka完成首次全合成以来,Smith、Comins、Micalizio和Cha也相继报道了新颖而简洁的合成路线。
Toyooka Group (2003)
2003年,日本富山医科药科大学的Toyooka小组率先合成天然205B的对映体,并指认了天然产物的绝对构型[1]。 从已知原料1出发,经过甲基铜锂的Michael加成,随后进行还原缩合、脱保护基和一系列氧化,再次得到α,β不饱和酯2。再次使用甲基铜锂进行Michael加成,得到单一的异构体3。作者推测铜锂试剂进攻的过程中A1,3空间静电作用是高选择性产生的原因[2]。在从上方进攻过程中,只受到Hc的排斥,而从下方进攻则会受到Ha和Hb两个原子的A1,3排斥作用。将3水解之后经过一些列简单转化得到不饱和醛4。
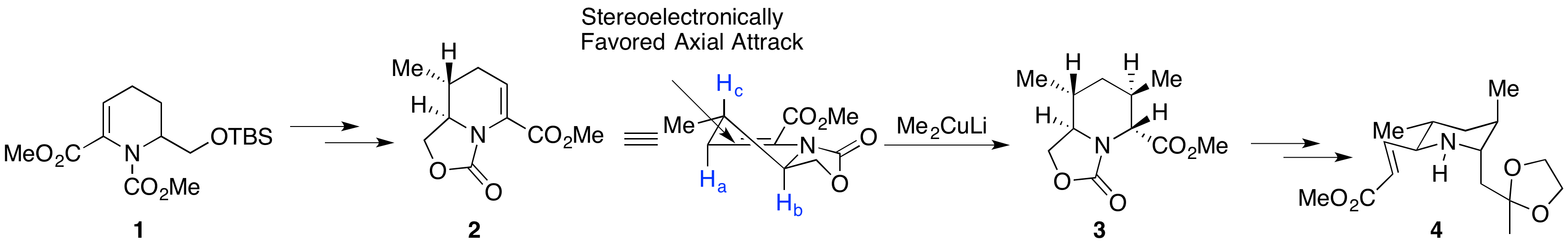
化合物4经过选择性还原和脱保护基之后得到5,在酸性条件下发生Mannich反应,一锅完成第二、第三个环的构建。而后发生Wittig反应,在酸性条件下发生双键异构得到立体选择性的(+)-205B。

关键反应:立体选择性Michael加成、Mannich反应
参考文献:
[1] Toyooka, N.; Fukutome, A.; Shinoda, H.; Nemoto, H. Angew. Chem., Int. Ed. 2003, 42, 3808.
[2] Hoffmann, R. W. Chem. Rev. 1989, 89, 1841.
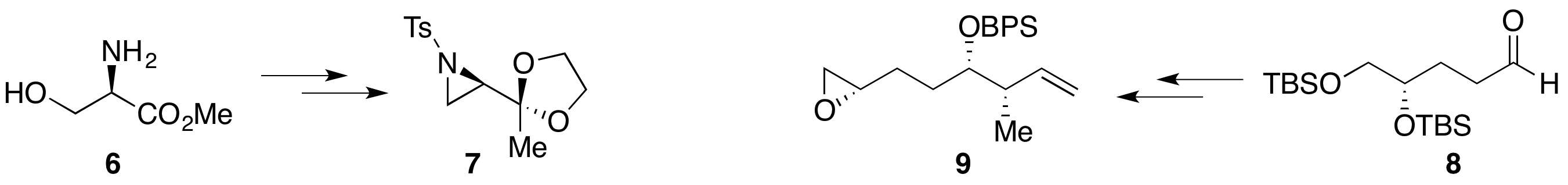
Smith Group (2005)
设计一个多组分反应考验了化学家对于反应机理的理解力和创造力。2005年,宾夕法尼亚大学的A. B. Smith, III利用三组分的阴离子接力化学(ARC)、一锅串联环化和烯烃关环复分解(RCM),用19步实现了(−)-205B的全合成[3]。
丝氨酸甲酯6经过Mitsnobu等反应转化为氮杂环丙烷缩酮7;已知物二羟基戊醛8通过Brown不对称巴豆基化、连接/脱除保护基和Fraser-Reid环氧化(TrisIm策略),得到高度非对映选择性的环氧化合物9。
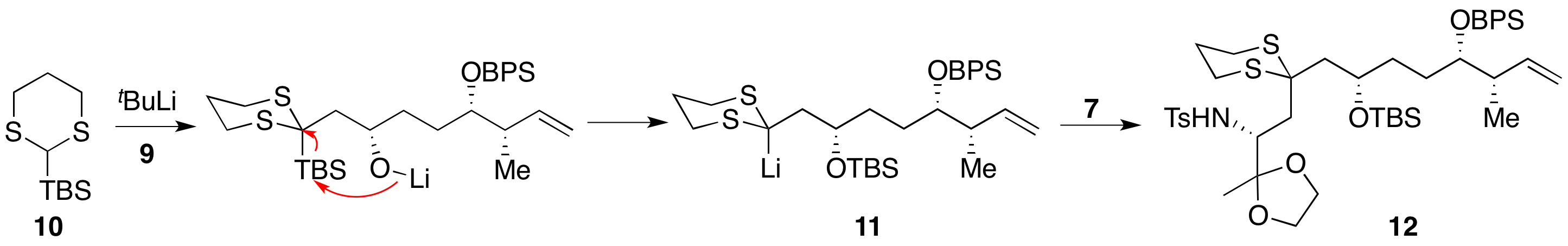
将二硫缩醛10在低温下锂化,随后与8反应开环。过程中形成的氧负离子进攻分子内的TBS基团,重新得到二硫稳定的碳负离子11。这时加入7,则会再次发生SN2开环反应,得到长碳链的三组分偶联产物12。该过程称为阴离子接力化学(Anion Relay Chemistry, ARC)[4],Smith许多全合成都是基于此方法发展而来。
脱除分子中的BPS保护基,使用MsCl酯化两个仲羟基,随后分别在碱性条件和钠汞齐脱除Ts保护基的条件下实现了两次分子内关环反应得到13。用酸水解缩酮保护基,并转化为烯醇硅醚,使用Grubbs二代催化剂实现分子内关环复分解得到三环骨架14。之后,利用Wittig反应引入亚甲基并调整氧化态即得到(−)-205B。
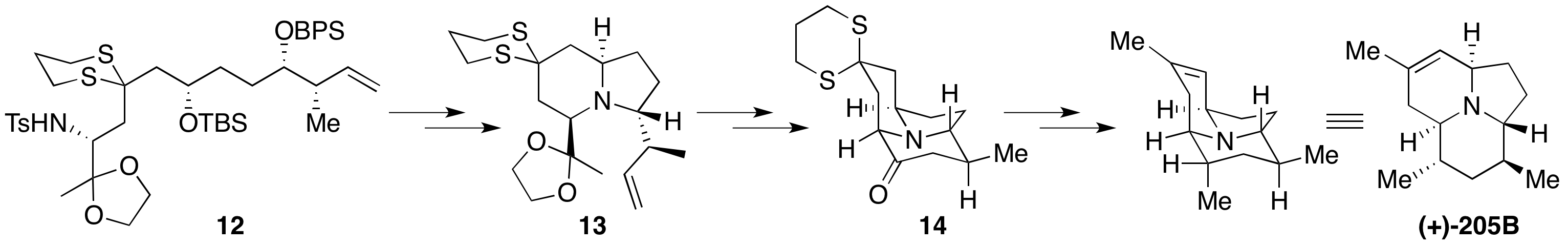
关键反应:阴离子接力化学、烯烃复分解
参考文献:
[3] (a) Smith, A. B.; Kim, D.-S. Org. Lett. 2005, 7, 3247. (b) Smith, A. B.; Kim, D.-S. J. Org. Chem. 2006, 71, 2547.
[4] Smith, A. B.; Xian, M. J. Am. Chem. Soc. 2006, 128, 66.
Comins Group (2011)
北卡罗莱纳州立大学的Comins教授从吡啶衍生物15出发,进过二氢吡啶酮中间体,以11步反应总收率8%得到(−)-205B。