初见GaussView
Gaussian软件是目前化学领域最流行、应用范围最广的量子化学计算程序包。它最早是由约翰·波普(John. A. Pople, 1998年诺贝尔化学奖获得者)在70年代开发的。Gaussian软件基于量子力学,致力于把量子力学理论应用于实际问题。同时Gaussian界面友好、参数多已经被设置好,大大降低了科研工作者从事量子力学的门槛,同时也是初学者利用量子力学理解化学的首选工具。GaussView是Gaussian软件默认的可视化工具,可以用来编辑输入文件以及打开输出文件。

本篇教程不涉及任何关于量子力学的内容,而仅仅说明GaussView的使用方法。同时也希望读者能将Gaussian当做一个用于理解化学的软件,任何对于量子化学的不解都不应成为使用Gaussian的障碍。
在我们打开了GaussView之后,出现了如下的界面:
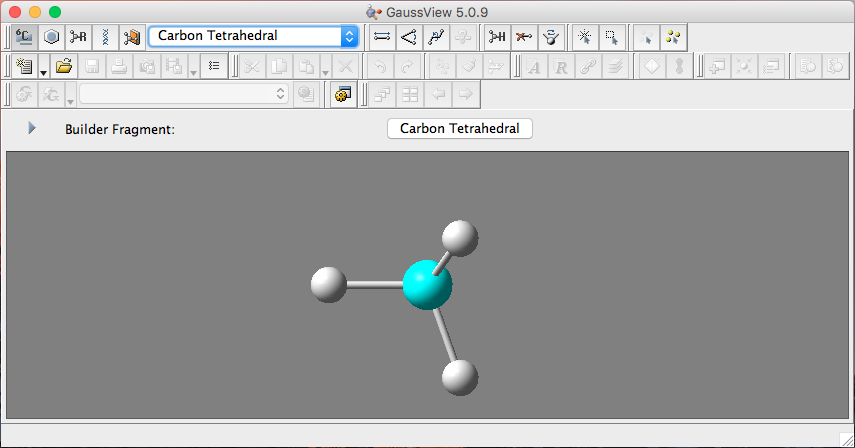 下方显示的是所要画的原子,高亮的是连接原子,图中所示代表甲基。
下方显示的是所要画的原子,高亮的是连接原子,图中所示代表甲基。
上方的各种工具栏各具功能,在用GaussView构建分子时,我们最常用到的是Builder工具栏:
 从左到右的图标依次是:元素、环、官能团、生物分子片段、自定义片段;键长调整、键角调整、二面角调整、参数测量;增加氢原子、删除原子、翻转原子基团;单击选中原子、框选原子、取消选中和全选。
从左到右的图标依次是:元素、环、官能团、生物分子片段、自定义片段;键长调整、键角调整、二面角调整、参数测量;增加氢原子、删除原子、翻转原子基团;单击选中原子、框选原子、取消选中和全选。
下面一段视频介绍如何利用GaussView中的功能构建复杂的有机分子。
我们发现,这样画出的分子在一些地方有些别扭,只需要单击Edit → Clean进行简单处理就完成了。
优化处理完成的效果:
使用Clean这种方法可以迅速的将所画的结构变得“赏心悦目”,但是这是相当经验的手段,得到的结构需要利用计算的手段做进一步优化。
后面的教程将建立在Exploring Chemistry with Electronic Structure Methods这本书的第三版案例之上,该书由Gaussian公司出版,官网为expchem3.com。最基础的理论背景已经在其官网上有所介绍,我们也会在日后进行整理与翻译的工作。
能量计算与结构优化
在我们完成分子的绘制之后,我们就需要对其进行能量的定量计算与几何结构优化。
首先,我们先了解一下计算的方法。我们最终的目的都是希望通过各种近似去找到Schrödinger方程的解,而不同的近似方法就对应了不同的精度,这些理论方法也称为姿势理论水平(Levels of Theory);一般来说,越高级的方法,对应着越高的精度,同时也会利用越大的计算资源。
Gaussian针对于不同大小的体系,可以选用不同的方法,如使用牛顿力学的分子力学方法(MM2、UFF)、半经验方法(PM6、AM1)、Hartree-Fock理论(HF)、Møller-Plesset微扰理论(MP2、MP4)、耦合簇理论(CCSD(T))、密度泛函理论DFT(B3LYP、APFD、M06、CAM-B3LYP)等等。当然,每一种方法都有使用范围和局限性。在教程中,主要涉及到有机化合物的优化,密度泛函理论中的B3LYP方法是我们解决此类问题的一般性选择。
计算模型下另一个需要的是基组(Basis Set)。基组是量子力学用来描述分子波函数的一系列数学函数。基组将电子限制在特定的空间区域之中,是由原子轨道的概念发展而来。常见的基组包括:最小基组,劈裂价键基组(极化基组,弥散基组),以及涉及到电子相关作用的高角动量基组。
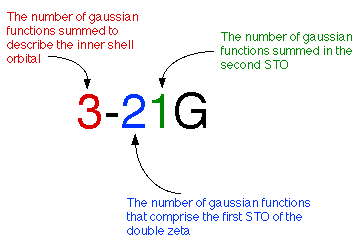
劈裂价键基组内层为单个Slater轨道,只对外层轨道计算double-zeta。

极化基组允许加入更高的轨道角动量,改变轨道形状。
参考资料:Wikipedia
在了解了这些知识之后我们就能对分子进行定量的计算。
首先在GaussView中绘制一个甲醛分子,然后右击,选择Calculate → Gaussian Calculation Setup。
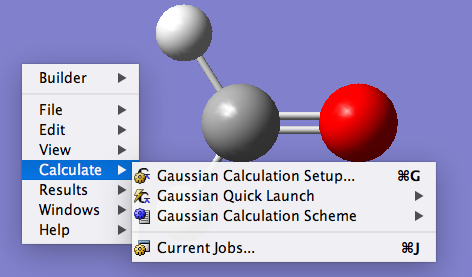
随之弹出设置计算参数的窗口。在任务类型(Job Type)中选择Optimization;
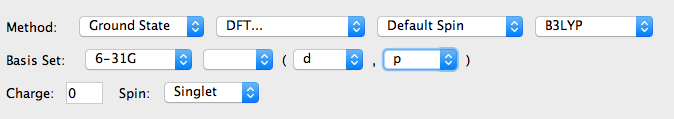
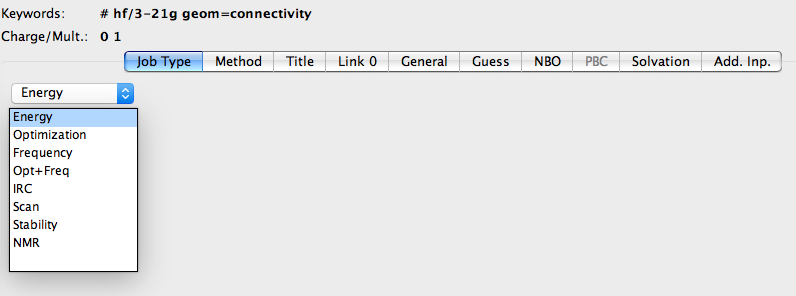 Method中选择DFT、B3LYP、6-31g(d,p),设置好正确的电荷(Charge)和自旋多重度(Spin);
Method中选择DFT、B3LYP、6-31g(d,p),设置好正确的电荷(Charge)和自旋多重度(Spin);
 在任务标题(Title)选项卡中输入计算的任务的名称,然后就可以点击Submit,保存输入文件并运行Gaussian了。
在任务标题(Title)选项卡中输入计算的任务的名称,然后就可以点击Submit,保存输入文件并运行Gaussian了。
用GaussView打开输出的结果,使用Inquire工具查看优化后的键长、键角和二面角。