Synthesis of Axinellamines
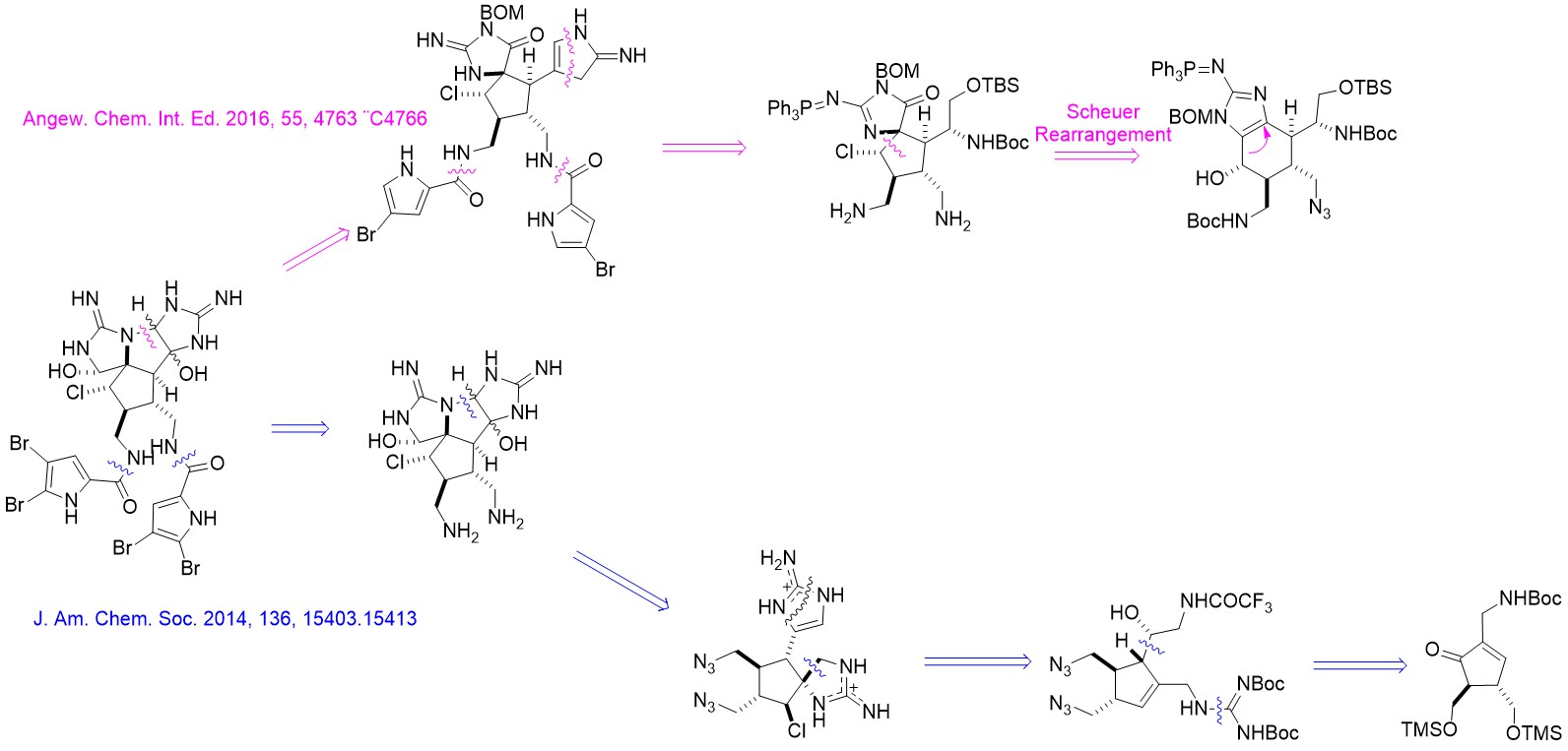
Axinellamines A&B是有着复杂多环骨架的广谱抗菌吡咯-咪唑生物碱。1999年由澳大利亚格里菲斯大学的Ronald J. Quinn等人从澳大利亚海绵中提取并进行了结构解析[1] 。该分子有诸多氮原子和卤原子(C:N:X ≈ 4:2:1),并有着有趣的五元环环系结构。2011年,Phil S. Baran对该分子完成首次全合成。在2016年,Chuo Chen基于对结构类似的天然产物Massadine及其衍生物的合成研究,提出了该天然产物的另一条合成路线。
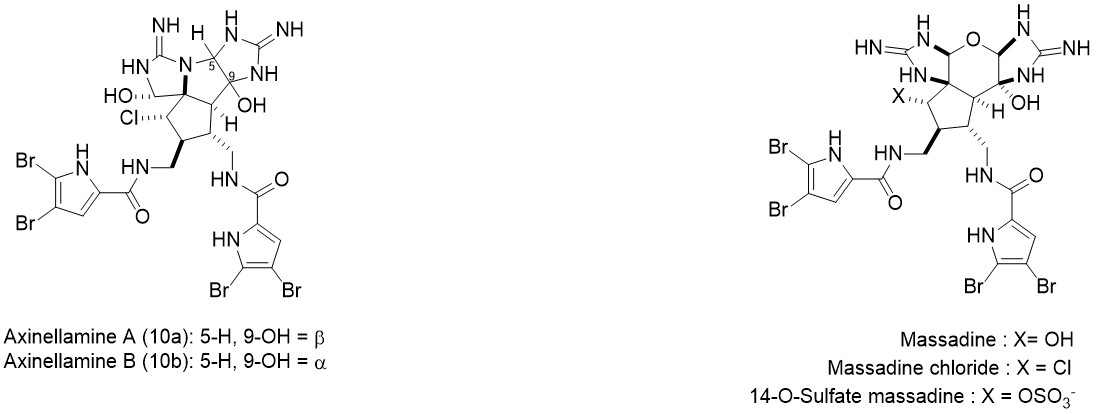
参考文献:
[1]S. Urban, P. A. Leone, et. al. J. Org. Chem. 1999, 64, 731.
图片来源:
Ma, Z.;Wang, X.;Ma, Y.; Chen, C. Angew. Chem. Int. Ed. 2016, 55, 4763.
Baran Group (2011&2014)
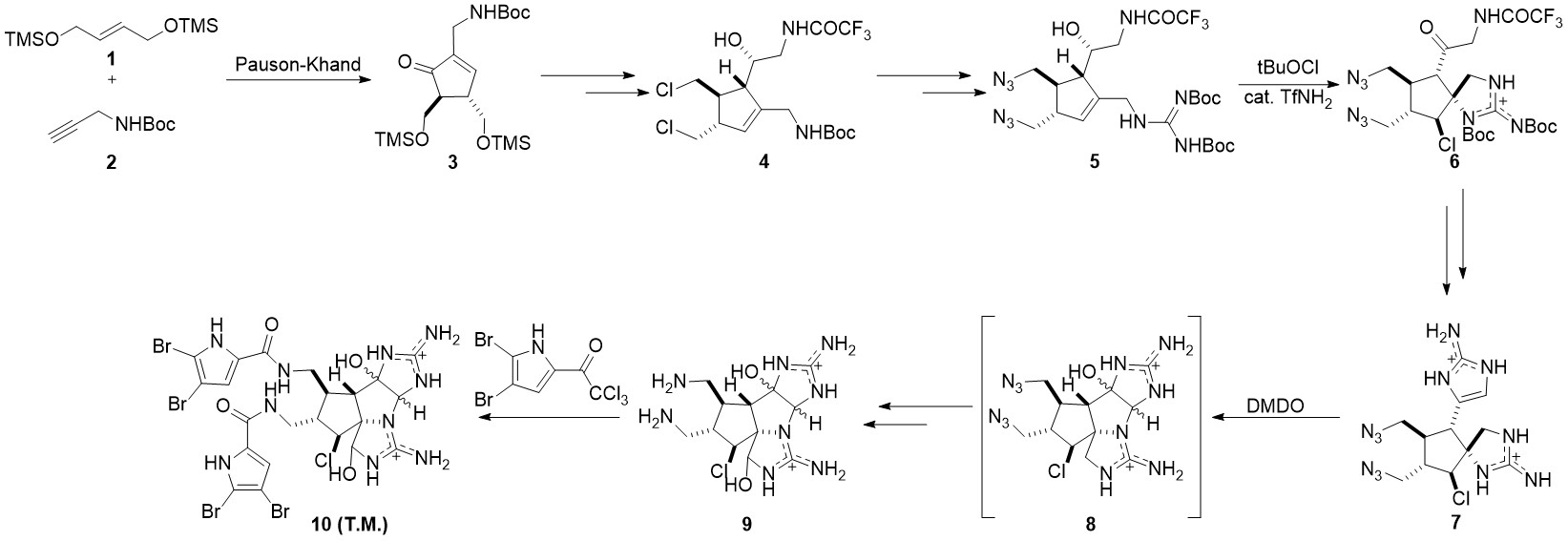
2011年,Phil Baran小组完成了该分子的全合成工作。该路线可用于执行克级(gram scale)合成[2]。该路线由较简单的有机小分子1和2出发,经过Pauson-Khand反应生成环戊烯酮3。随后经过脱保护基,氯代,并经过负离子化后和醛发生亲核加成形成中间体4。4经过一系列取代反应生成胍5。构筑中间物6螺环的步骤在多次条件筛选后选用以tBuOCl为氯化试剂,TfNH2为催化剂的条件实现。经过脱保护,氰胺环化后,中间物7中另一个五元氮杂环被成功构筑。在DMDO的作用下,分子7实现单步羟基化与环化,转化为中间物8。在羟化,叠氮基还原,酯化后最终生成目标产物10。
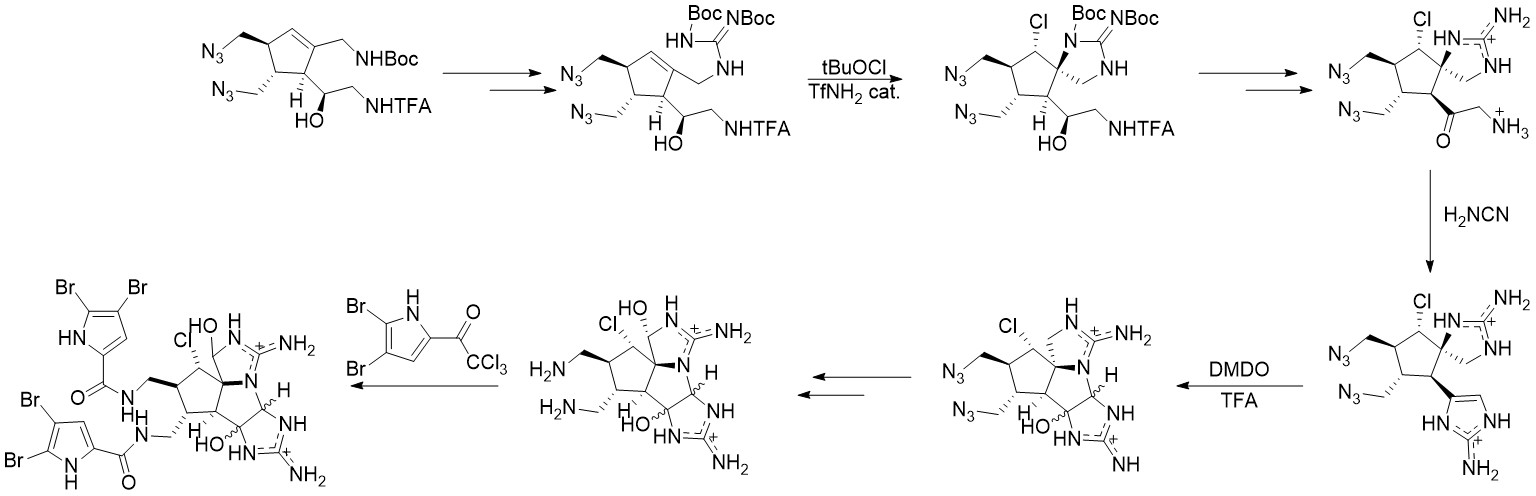
通过不同的逆合成策略,Baran小组提出了另一条路线[3]。该路线关键反应选择与Baran于11年的路线选择相似,在此仅做路线展示,不做过多描述。
#####关键反应:Pauson-Khand反应 烯烃亲电加成
#####参考文献:
[2]Su, S.; R. A. Rodriguez; P. S. Baran J. Am. Chem. Soc. 2011, 133, 13922–13925
[3]R. A. Rodriguez; D. B. Steed; Y. Kawamata, Su, S.; P. A. Smith; T. C. Steed; F. E. Romesberg; P. S. Baran J. Am. Chem. Soc. 2014, 136, 15403−15413
###Chen Group (2016)
在2016年,德克萨斯大学的Chen小组基于已经合成的前体11[4]设计了另一条路线[5]。该路线可形成单一构型的目标产物10b
前体11经过两步基本转换成为中间体12,随后经过Scheuer重排完成螺环骨架构筑,转换为中间体13。之后经过氯代,脱保护,还原等步骤生成中间体14。需要注意的是,还原的手性是由于邻基参与过程决定的,脱保护是选择脱除位阻较小的氨基上的Boc保护基。14经过酯化,氧化,以及与Baran组相似的氰胺环化后形成中间体15。15经过SmI2单电子还原后,吡咯alpha位的溴脱落,三苯基膦脱落,形成中间体16。用AD-mix-α[6]氧化环化后生成17,再经过后续的脱保护,溴化可形成单一构型的目标产物10b。作者也尝试过先环化后还原的策略,该过程生成了10a和10b的混合物。
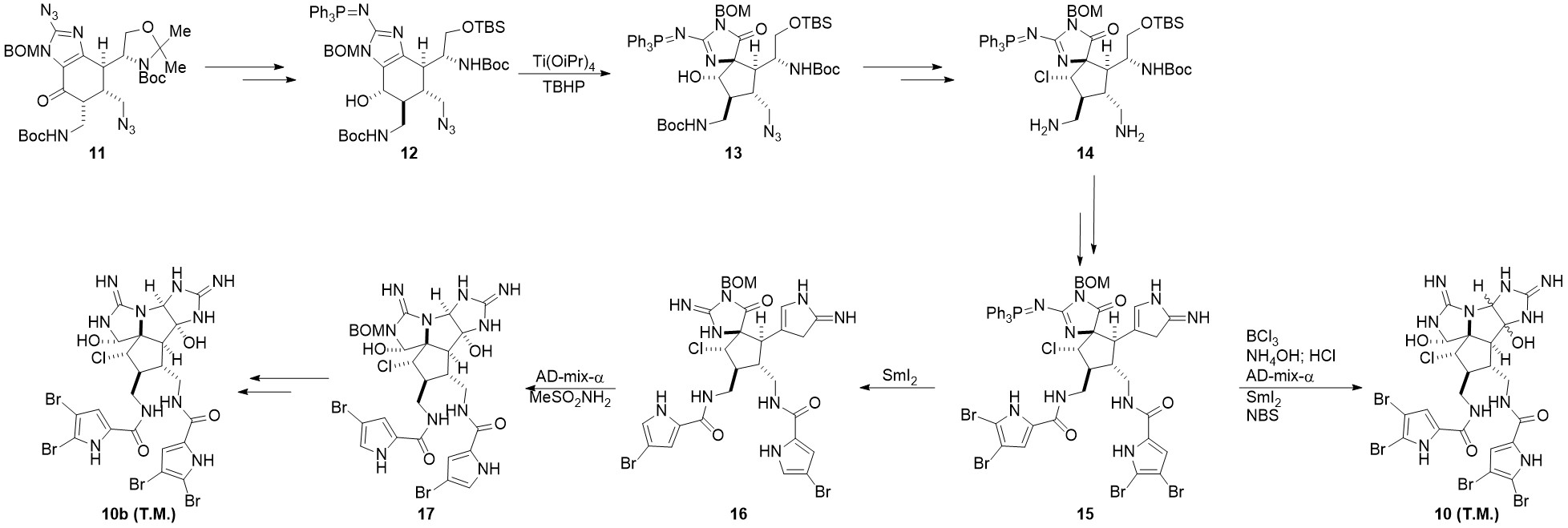
#####关键反应:Scheuer重排
参考文献:
[4]X. Tan, C. Chen, Angew. Chem. Int. Ed. 2006, 45, 4345–4348
[5] Z. Ma, X. Wang, Y. Ma, C. Chen, Angew. Chem. Int. Ed. 2016, 55, 4763 –4766
[6]https://en.wikipedia.org/wiki/AD-mix