MRCC是由匈牙利布达佩斯大学开发的主攻高精度量化计算的软件包,其亮点在于可用于任意阶耦合簇(包括其各种微扰近似)和组态相互作用,并有对应的解析梯度。
笔者将对该软件的耦合簇功能进行试用。限于笔者的电脑的计算力(Xeon E5 2696V3),CC只算到三阶;引入微扰近似后只算到四阶,即最高计算水平为CCSDT(Q).
使用水分子和羟基负离子用于测试,结构在M06-2X/def2-TZVP下优化得到。
MRCC程序的输入文件和操作流程都比较独特。输入文件在提交计算时必须改名成MINP(必须大写)才可计算,对应的命令为dmrcc > xxx (xxx为输出文件的名称)
输入文件格式如下图:
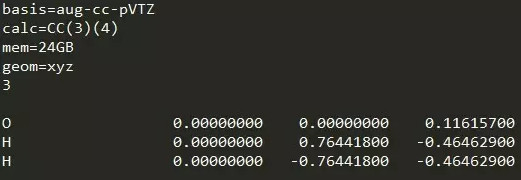
CC(3)(4)对应CCSDT(Q)。这样计算同时会输出CCSDT[Q]的结果。
虽然官方手册上写并行计算需要使用MPI并有一些特殊操作,但亲测提交任务后自动并行占满全部CPU.
用aug-cc-pVTZ基组,测试了水分子和羟基负离子在HF, CCS(D), CCSD, CCSD(T), CCSD[T], CCSDT, CCSDT(Q), CCSDT[Q]下的能量(CCSDTQ或更高阶的耗时已经不可接受)。
输出文件是这个画风:
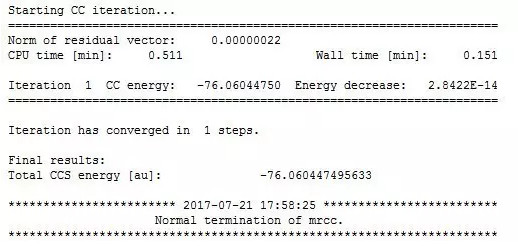
然后是相关能,以CCSDT(Q)结果作为标准:
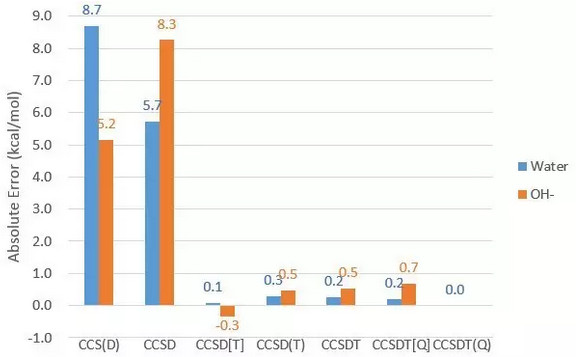
低于三阶的耦合簇误差都相当大,而CCSD(T)的结果和CCSDT相当甚至更好。进一步加到四阶后并没有显著改进。
同时还把MRCC的CCSD(T)结果与Gaussian相同条件下的结果进行了对比,几乎相同(区别在于输出值的最后两位)。
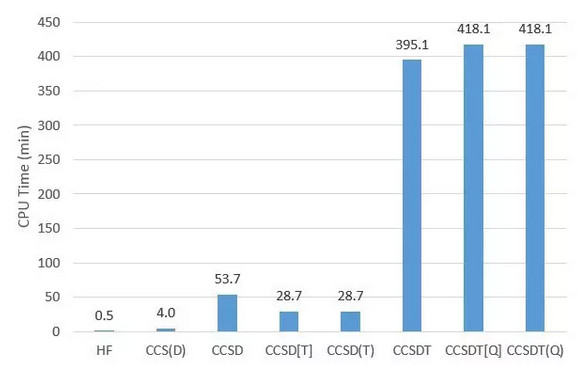
上图是MRCC程序实现的不同方法的CPU时间比较。可以看到引入第n阶的微扰近似后耗时与无近似的(n-1)阶CC相同,而精度几乎没有损失。其中CCSD的耗时高于CCSD(T)是由于内存设置的原因,可以无视。
同时还检查了羟基负离子结合质子生成水的气相反应能(只考虑电子能量),仍然以CCSDT(Q)结果为基准:
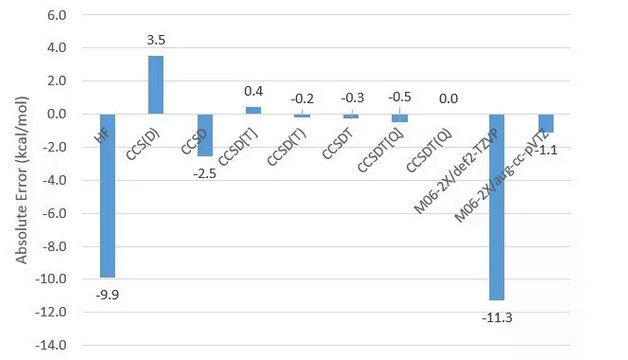
其中DFT方法用Gaussian实现。M06-2X在相同基组下表现相当好。由于这个反应涉及阴离子,因此M06-2X/def2-TZVP表现不太好也是可以理解的;尽管如此,误差这么大甚至差于HF还是令人意外。
同时还做了一个测试,把构型优化水平改到B3LYP/6-31G*,以刚才M06-2X/def2-TZVP构型得到的CCSDT(Q)反应热为标准,观察各种CC和DFT水平算出的结果的误差。
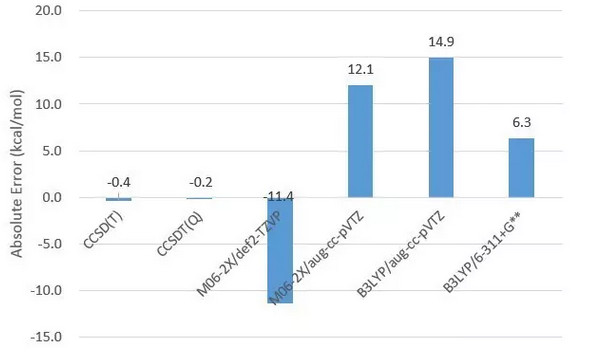
CCSD(T)和CCSDT(Q)的结果和之前几乎相同,而之前表现较好的M06-2X/aug-cc-pVTZ的误差则大得惊人。令人惊讶的是此时采用较小的基组的DFT水平(如B3LYP/6-311+G**)反而误差较小。这从一定程度上说明对于低水平的构型优化,单点能计算可能未必水平越高越好,而是存在微妙的误差抵消现象。 最后比较一下MRCC和Gaussian的运行效率。全部采用36核心(18物理+18超线程)时,水分子的CCSD(T)单点,MRCC总CPU时间高达28.7min, 而Gaussian仅仅3min左右。说明MRCC程序其实在性能上并没有任何优势。 最后,由于本文中只测试了一个体系,因此对诸多方法精度等的测试很难说有普遍意义,需要谨慎采信。